Olika typer av neurofibromatos
Det finns tre varianter av neurofibromatos: typ 1 (von Recklinghausens sjukdom), typ 2 och typ 3 (schwannomatos). Symtomen och sjukdomsförloppet skiljer sig åt mellan de olika varianterna. Alla tre formerna är kroniska. Ungefär hälften av personerna med NF typ 1 har plexiforma neurofibrom, vilket betyder att man har medfödda tumörer som växer längs större nerver och kan orsaka smärta och högre risk för elakartade tumörer.
Symtom på neurofibromatos
Symtomen vid neurofibromatos är mycket olika hos olika personer, till och med hos de inom samma familj som drabbas. De flesta får symtom redan som barn. Neurofibromatos kan bl a visa sig genom symtom i huden, ögonen, skelettet och nervvävnaden, och besvären kan dessutom variera kraftigt i omfattning.
Neurofibromatos typ 1:
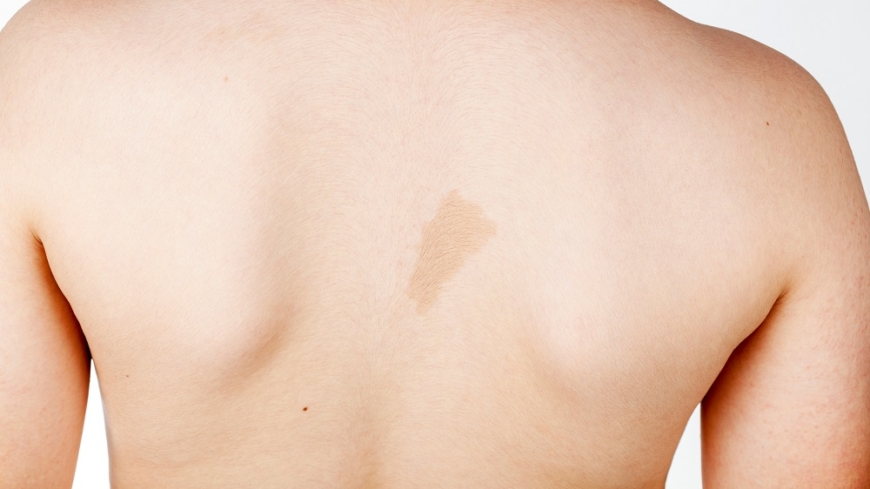
- Café au lait-fläckar (släta, oregelbundna fläckar på huden)
- Fräknighet i armhålor och ljumskar
- Neurofibrom (små hudtumörer)
- Språksvårigheter
- Inlärningssvårigheter
- ADHD-liknande symtom
Neurofibromatos typ 2:
- Problem med hörseln (nedsatt hörsel eller dövhet)
- Yrsel
- Balansproblem
Den underliggande orsaken till dessa symtom hos personer med NF-2 är godartade tumörer i hjärnhinnorna och centrala nervsystemet. Symtomen vid typ 3/schwannomatos liknar de vid typ 1, och brukar domineras av smärta. Ungefär hälften av de med neurofibromatos har inga allvarliga symtom – det enda tecknet är så kallade café au laitfläckar (CAL).
Orsaker till neurofibromatos
Neurofibromatos orsakas av muterade gener som förs vidare. Den största riskfaktorn för att drabbas är att sjukdomen finns inom familjen. De olika typerna av neurofibromatos påverkar olika kromosomer. NF typ 1 och 2 förs vidare från föräldrar genom s k dominanta anlag. Det innebär att ett barn med en förälder med NF 1 eller NF 2 har 50 % chans att ärva mutationen. Ärftligheten vid NF typ 3 är däremot mer oklar.
NF typ 1 beror på sjukdomsframkallande förändringar i NF1-genen, som finns på den långa armen av kromosom 17. Denna gen ansvarar för att koda för proteinet neurofibromin, som tjänar som tumörhämmare.
Undersökning och diagnos
Om det finns tecken på neurofibromatos är det viktigt att bli undersökt av en läkare, oftast en neurolog. Det kan behövas olika undersökningar som t ex datortomografi, magnetkamera och ultraljud. Ett gentest kan bekräfta diagnosen. Om någon i familjen får diagnosen kan övriga familjemedlemmar behöva kontrolleras.
Behandling av neurofibromatos
Neurofibromatos är en sjukdom som inte går att bota, men ibland kan tumörer avlägsnas, t ex de som ligger nära hörselnerven hos personer med NF typ 2 och riskerar att hota hörseln. Det kan också handla om tumörer som trycker emot nerver och leder till smärta. Tumörer behandlas på olika sätt beroende på dess storlek, typ, stadie och placering i kroppen. Neurofibrom i huden kan tas bort, dock riskerar de att växa tillbaka igen. Behandlingen fokuserar vanligtvis på att lindra symtom och kompensera för de funktionsnedsättningar som sjukdomen eventuellt ger.
Barn med NF typ 1 bör ha årlig uppföljning upp till tio års ålder av en barnläkare och ögonläkare och därefter vartannat år. Vid 16-18 års ålder rekommenderas magnetkameraundersökning (MR) av hela kroppen för att upptäcka djupt sittande neurofibrom och vid behov följa tumörutvecklingen. Vuxna med NF typ 1 följs av specialister och primärvården.
Forskning för att hitta en effektiv medicinsk behandlingsform är pågående. Även psykologiskt stöd kan behövas, eftersom diagnosen kan innebära en stor livsomställning.
Prognos
Livet med neurofibromatos ser ganska olika ut för olika personer som drabbas, eftersom symtomen varierar kraftigt i omfattning och karaktär. En del kan leva ett i princip normalt liv, medan andra behöver viss eller stor hjälp. Det brukar dock behövas regelbundna läkarkontroller under livets gång, t ex för att kontrollera blodtrycket.
När bör du söka vård?
Ju tidigare neurofibromatos upptäcks, desto större är möjligheterna att bromsa sjukdomens utveckling. Om du får symtom som kan tänkas bero på NF bör du söka vård på en vårdcentral.